
GitHub
Shendure Lab GitHub
Public GitHub repositories for assorted lab projects.
other software
Xpresso: Predicting gene expression levels from DNA sequences
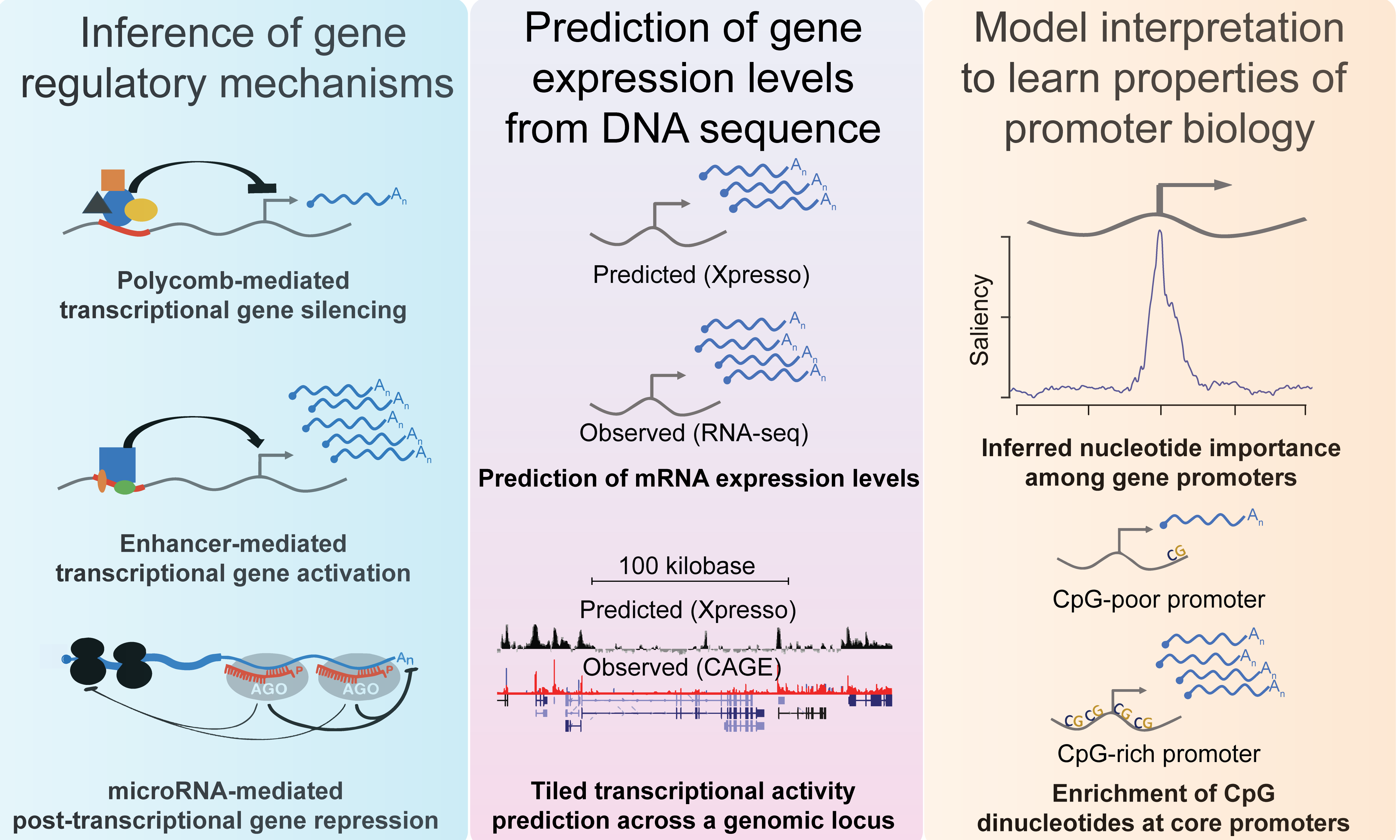
Xpresso is a software suite whose goal is to predict gene expression levels and transcriptional activity from genomic sequences. It is trained using convolutional neural networks. Pre-trained models are available for the human, mouse, and several cell types for these species.
Further information about Xpresso, including links to data and source code, are available at https://github.com/vagarwal87/Xpresso.
Our manuscript describing Xpresso was published as: Agarwal V.*, Shendure J.* Predicting mRNA abundance directly from genomic sequence using deep convolutional neural networks. Cell Rep. 2020 May 19;31(7):107663. doi: 10.1016/j.celrep.2020.107663. PubMed PMID: 32433972 .
Genome Editing of Synthetic Target Arrays for Lineage Tracing (GESTALT)
GESTALT is a method that uses genome editing to progressively introduce and accumulate diverse mutations in a DNA barcode over multiple rounds of cell division, thereby recording cell lineage relationships in the patterns of mutations shared between cells.
Further information about GESTALT, including links to data, reagents, source code, and phylogenetic trees for cell lineages of embryo and adult zebrafish, are available at http://gestalt.gs.washington.edu.
Our manuscript describing GESTALT was published as: McKenna A*, Findlay GM*, Gagnon JA*, Horwitz MS, Schier AF#, Shendure J#. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 2016 Jul 29;353(6298):aaf7907. doi: 10.1126/science.aaf7907. PubMed PMID: 27229144 .
Combined Annotation Dependent Depletion (CADD)
CADD is method that objectively weights and integrates diverse genomic annotations to a single, phred-scaled metric.
Further information about CADD, pre-computed CADD-based scores (C-scores) for all 8.6 billion possible single nucleotide variants (SNVs) of the human reference genome, and a tool for scoring of short insertions/deletions are available at http://cadd.gs.washington.edu.
Our manuscript describing CADD was published as: Kircher M*, Witten DM*, Jain P, O'Roak BJ, Cooper GM#, Shendure J#. A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics 2014 Mar;46(3):310-5. doi: 10.1038/ng.2892. PubMed PMID: 24487276.
Optimized Modeling and Design of Molecular Inversion Probes for Targeted Resequencing (MIPgen)
Molecular inversion probes (MIPs) enable cost-effective multiplex targeted gene resequencing in large cohorts. However, the design of individual MIPs is a critical parameter governing the performance of this technology with respect to capture uniformity and specificity. MIPgen is a user-friendly package that simplifies the process of designing custom MIP assays to arbitrary targets. New logistic and SVM-derived models enable in silico predictions of assay success, and assay redesign exhibits improved coverage uniformity relative to previous methods, which in turn improves the utility of MIPs for cost-effective targeted sequencing for candidate gene validation and for diagnostic sequencing in a clinical setting.
MIPgen is available for non-commercial use at: http://shendurelab.github.io/MIPGEN.
Our manuscript describing MIPgen was published as: Boyle EA#, O'Roak BJ, Martin BK, Kumar A, Shendure J#. MIPgen: optimized modeling and design of molecular inversion probes for targeted resequencing. Bioinformatics. 2014 Sep 15;30(18):2670-2. doi: 10.1093/bioinformatics/btu353. PubMed PMID: 24867941.
Genome Assembly with Hi-C-based Contact Probability Maps (LACHESIS)
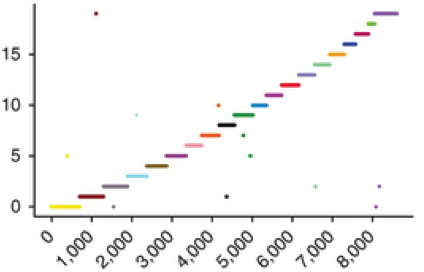
LACHESIS is method that exploits contact probability map data (e.g. from Hi-C) for chromosome-scale de novo genome assembly.
Further information about LACHESIS, including source code, documentation and a user's guide are available at: http://shendurelab.github.io/LACHESIS.
Our manuscript describing LACHESIS was published as: Burton JN#, Adey A, Patwardhan RP, Qiu R, Kitzman JO, Shendure J#. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature Biotechnology 2013 Dec;31(12):1119-25. doi: 10.1038/nbt.272. PubMed PMID: 24185095.
Genome Assembly with Contiguity Preserving Transposition (fragScaff)
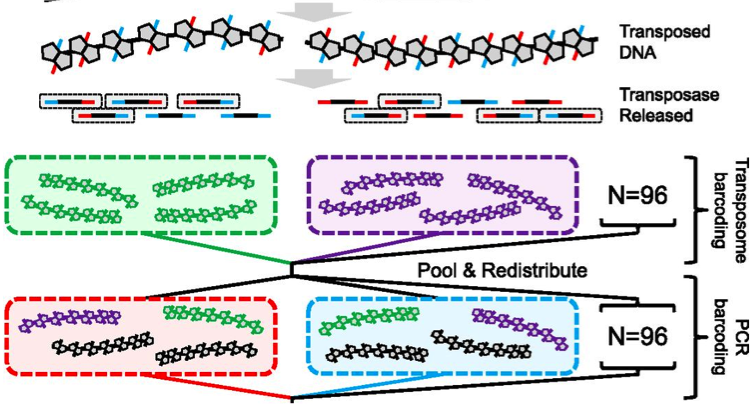
Contiguity preserving transposition and sequencing (CPT-seq) is an entirely in vitro means of generating libraries comprised of 9216 indexed pools, each of which contains thousands of sparsely sequenced long fragments ranging from 5 kilobases to >1 megabase. This software, fragScaff, leverages coincidences between the content of different pools as a source of contiguity information for scaffolding de novo genome assemblies. FragScaff is complementary to Lachesis, providing midrange contiguity to support robust, accurate chromosome-scale de novo genome assemblies without the need for laborious in vivo cloning steps.
Further information about fragScaff, including source code, is available at: https://sourceforge.net/projects/fragscaff/files.
Our manuscript describing fragScaff was published as: Adey A, Kitzman JO, Burton JN, Daza R, Kumar A, Christiansen L, Ronaghi M, Amini S, L Gunderson K, Steemers FJ, Shendure J#. In vitro, long-range sequence information for de novo genome assembly via transposase contiguity. Genome Research 2014 Dec;24(12):2041-9. doi: 10.1101/gr.178319.114. PubMed PMID: 25327137.
Species-level Deconvolution of Metagenome Assemblies with Hi-C-based Contact Probability Maps (MetaPhase)
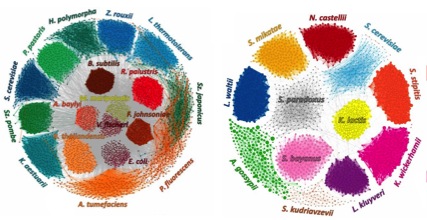
Microbial communities consist of mixed populations of organisms, including unknown species in unknown abundances. These communities are often studied through metagenomic shotgun sequencing, but standard library construction methods remove long-range contiguity information; thus, shotgun sequencing and de novo assembly of a metagenome typically yield a collection of contigs that cannot readily be grouped by species. MetaPhase is software that exploits chromatin-level contact probability maps, e.g., as generated by the Hi-C method, to reconstruct the individual genomes of microbial species present within a mixed sample.
Further information about MetaPhase, including source code, documentation and a user's guide are available at: https://github.com/shendurelab/MetaPhase.
Our manuscript describing MetaPhase was published as: Burton JN*, Liachko I*, Dunham MJ#, Shendure J#. Species-level deconvolution of metagenome assemblies with Hi-C-based contact probability maps. G3 (Bethesda). 2014 May 22;4(7):1339-46. doi: 10.1534/g3.114.011825. PubMed PMID: 24855317.